

A patogénese e as lesões causadas pela infecção por PRRSV dependem muito das características comuns entre os vírus pertencentes ao género Arterivirus: (i) capacidade de entrar e replicar-se nas suas células alvo, os macrófagos, (ii) virémia prolongada após a infecção, (iii) infecção persistente de longa duração (não permanente). A fase aguda da virémia nos porcos de engorda dura cerca de 4 semanas, após este periodo o vírus encontra-se, predominantemente, no tecido linfóide – normalmente durante o resto da sua vida para um animal de produção – e periodicamente a virémia pode reproduzir-se (revisto em detalhe em Chand et al. 2012, Curr Opin Virol).
As lesões induzidas pelos vírus respiratórios no tecido pulmonar podem desenvolver-se por 2 vias principais: desregulação por destruição directa dos pneumócitos e/ou infecção das células inflamatórias e imunitárias que induzem o dano tissular através da libertação de citoquinas activas ou metabolitos nocivos. Estas células activadas podem recrutar outras células inflamatórias para o local da infecção aumentando o dano tissular, mas também têm um papel importante na eliminação viral e regulação da inflamação.

As células alvo nas infecções por PRRSV são os macrófagos alveolares. O dano tissular é a consequência da apoptose directa (e necrose) de ditas células e – em maior medida – as suas células vizinhas (efeito by stander) devido à libertação de citoquinas apoptogénicas, espécies reactivas de oxigénio e óxido nítrico. Ao mesmo tempo libertam citoquinas pró-inflamatórias, que são responsáveis pelo recrutamento de outras células inflamatórias e algumas delas causam os sintomas sistémicos (febre, letargia, etc). Também são segregadas citoquinas anti-inflamatórias e reguladoras. Está bem documentado que, do mesmo modo que para outros vírus, o PRRSV tem a capacidade de suprimir a resposta primária do interferón tipo I – um potente mecanismo anti-viral da imunidade inata – nas primeiras fases da infecção através das suas proteínas não estruturais, o que lhe permite replicar-se e disseminar-se de um modo mais eficiente. No entanto, diferentes isolados de PRRSV têm diferentes capacidades imunomoduladoras pelo que os sinais clínicos, as lesões e o resultado global da infecção dependem muito do peso dos mecanismos que acabam de ser descritos. O dano/destruição dos macrófagos pode facilitar o aparecimento de infecções secundárias como costuma ser observado em condições de campo.
Nas infecções experimentais realizadas em explorações isoladas, os sinais clínicos e as lesões observadas dependem muito da patogenicidade da estirpe utilizada para inocular os animais. As lesões respiratórias mais severas observam-se aos 7–14 dias PI. Macroscopicamente trata-se de áreas consolidadas, escuras e salpicadas que afectam mais gravemente os lóbulos craneo-ventrais, mas que podem ser encontradas disseminadas em todo o tecido pulmonar (foto 1). As principais lesões histopatológicas incluem: (1) hipertrofia e hiperplasia pneumocitárias, (2) infiltração mononuclear septal, (3) restos necróticos intra-alveolares, (4) acumulação intra-alveolar de células inflamatórias e (5) acumulação perivascular de células inflamatórias.
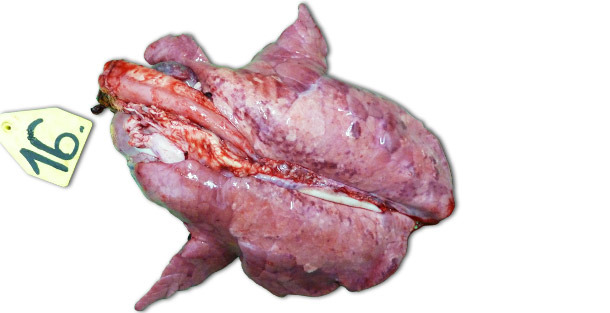
Foto 1. Pulmões de um leitão abatido 14 dias PI, inoculado com uma estirpe de alta patogenicidade tipo I subtipo 3 "Lena".
Num estudo com desafio no qual os porcos foram eutanasiados aos 14 e 21 dias PI, nos animais infectados foi observada uma diminuição significativa dos restos necróticos intra-alveolares e de células inflamatórias intra-alveolares aos 21 dias PI, por outro lado, foi observada apenas uma pequena alteração nas outras três categorias. Estas descobertas podem ser explicadas pelo facto de que a formação de restos necróticos intra-alveolares e a conseguinte acumulação intra-alveolar de células inflamatórias (basicamente granulócitos neutrófilos) refletem a fase aguda da doença (foto 2), na qual as substâncias nocivas libertadas pelos macrófagos infectados causam um grande dano tissular. Após a fase inicial e aguda – na ausência de infecções bacterianas secundárias – o sistema imunitário e os mecanismos naturais de cura eliminam o tecido necrótico pelo que as células da inflamação aguda (granulócitos neutrófilos) desaparecem, os alvéolos esvaziam-se e os pneumócitos danificados são substituídos por pneumócitos proliferativos tipo II.
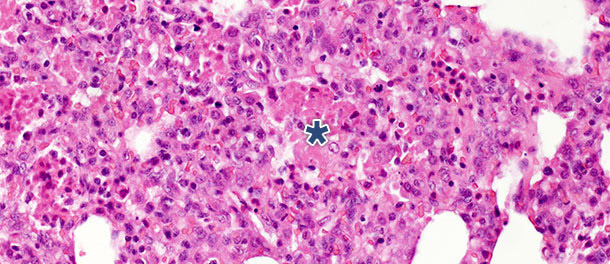
Foto 2. Restos necróticos intra-alveolares e acumulação de células inflamatórias (asterisco) no tecido pulmonar
aos 10 dias PI com um isolado de PRRSV tipo 1 sub-tipo 1. H.E. 200×
Os pneumócitos tipo II são células cuboidais que tendem a estar localizadas na inserção dos septos alveolares. No tecido pulmonar dos mamíferos constituem 60% de todas as células alveolares epiteliais, mas apenas cobrem cerca de 5% da superfície alveolar. O seu papel mais importante é a síntese, secreção e reciclagem do surfactante pulmonar, que reduz a tensão superficial alveolar, prevenindo o colapso durante a expiração. A outra função importante dos pneumócitos tipo II radica no seu potencial proliferativo. Quando os pneumócitos tipo I são danificados, os tipo II actuam como células progenitoras para os substituir e, com o tempo, diferenciar-se em pneumócitos tipo I. No desafio descrito mais acima, o número de ditas células aumentou significativamente nos 10 dias PI (foto 3) e não diminuiu aos 21 dias PI. Ainda que o estudo tenha terminado aos 21 dias PI e não tenha sido observada uma cura histológica total (restitutio ad integrum) no grupo infectado, os animais recuperaram da doença clínica e o elevado número de pneumócitos tipo II após a inoculação, tanto nas fases iniciais como finais da doença, demonstram a sua importância na cura do tecido pulmonar (descrito com detalhe em Balka et al. 2013. J Comp Pathol).
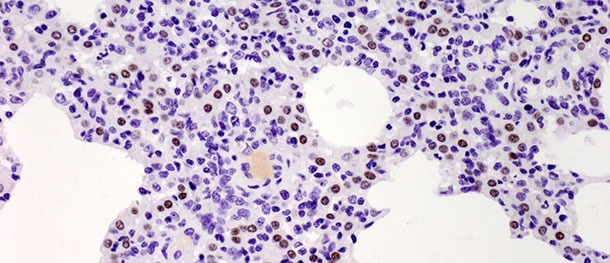
Foto 3. Os pontos castanhos indicam o núcleo dos pneumócitos tipo II identificados com anticorpos anti-TTF-1. Foi observado um aumento significativo no número de células positivas aos 10 dias após a infecção com um isolado tipo I sub-tipo 1. IHC 200×




