

Os desafios - não tão novos - quando se fala do vírus da PRRS
Não é novidade que o vírus da PRRS tem características importantes que contribuem para o reaparecimento constante da doença nas explorações de suínos, levando a problemas de saúde animal e perdas económicas consideráveis. Uma dessas características é a sua rápida capacidade de sofrer mutações e tornar-se potencialmente mais grave e/ou mesmo infectar animais anteriormente "imunes" a diferentes variantes da PRRS. Por esta razão, a fim de conhecer o "tipo" de vPRRS presente numa exploração - se se trata de um novo vírus de surto ou de um vírus "residente" - o genoma do vPRRS é frequentemente analisado.
Esta tarefa é geralmente efectuada através da sequenciação de uma parte importante do vPRRS chamada gene ORF5; para compreender onde o vírus se enquadra evolutivamente e como se compara com vírus previamente detectados na exploração ou nas proximidades/região.

A informação fornecida pela sequenciação do vPRRS tem sido amplamente utilizada em campo para:
- compreender qual o vírus que está a causar problemas
- obter orientações sobre a origem do surto
- para distinguir entre estirpes vacinais e estirpes "residentes" ou recém-introduzidas
- para a seleção da vacina, etc.
Recentemente, a classificação por "linhagem" tem sido cada vez mais utilizada para classificar os vírus. Este método foi desenvolvido por volta de 2010 (Shi et al., 2010), altura em que os vPRRS foram divididos em 9 linhagens diferentes; foi posteriormente aperfeiçoado quando foram subdivididos em sub-linhagens.
Uma verdade desconfortável: a sequenciação de uma amostra é suficiente?
A sequenciação da PRRS não é normalmente barata, pelo que é geralmente efectuada em "situações especiais" e não numa base de rotina (por exemplo, durante novos surtos, após casos de inoculação do vírus, etc.). Além disso, nestas situações, na grande maioria dos casos estudados para diagnóstico, apenas uma amostra ou um conjunto de amostras é utilizado para a sequenciação, de entre as muitas amostras normalmente colhidas na exploração.
A consequência é que, em muitos casos, os veterinários, os produtores e outros profissionais da saúde animal frequentemente utilizam a informação de uma única sequência para tirar conclusões importantes relativamente à origem do vírus (especialmente em novos surtos), orientar as investigações nos surtos e informar sobre futuras intervenções.
No nosso estudo, queríamos confirmar que, se sequenciássemos várias amostras recolhidas no mesmo dia, todas elas forneceriam a mesma sequência, ou sequências "suficientemente próximas" para serem consideradas, pelo menos, da mesma linhagem de vírus. Também queríamos experimentar a sequenciação do genoma completo (WGS) juntamente com a sequenciação da ORF5 para ver se conseguíamos obter mais informações. A WGS pode ter a vantagem de fornecer informações sobre todo o vírus, ao passo que a ORF5 tem sido tradicionalmente utilizada, mas apenas nos conta uma parte da história. No entanto, como se trata de um desenvolvimento relativamente recente e pode ser dispendioso (nos EUA custa cerca de três vezes mais do que a sequenciação da ORF5), ainda estamos a aprender a interpretá-lo e a nossa "base de dados" para comparação é muito mais pequena do que a da ORF5.
Suspeitámos que, dada a rápida taxa de mutação da PRRS e a grande dimensão das explorações suinícolas modernas, poderíamos encontrar várias linhagens de PRRS num único evento de amostragem, o que seria preocupante. Um resumo gráfico do raciocínio subjacente ao nosso estudo é apresentado na Fig. 1.
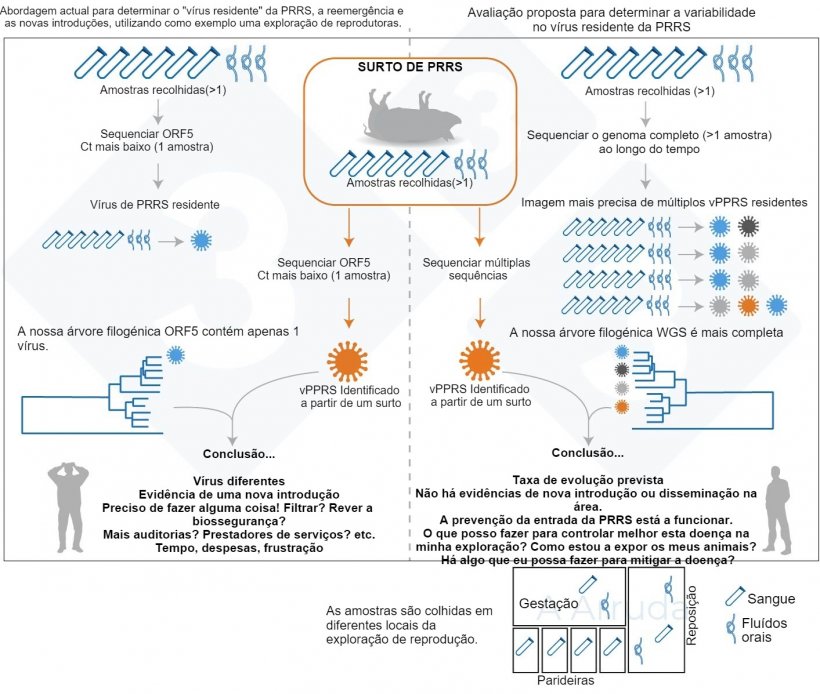
Investigar o número de estirpes de PRRS que se podem encontrar nas nossas explorações
Incluimos cinco explorações no estudo, 3 explorações de reprodutoras e 2 explorações de engorda; e recolhemos amostras mensalmente durante aproximadamente um ano, utilizando diferentes tipos de amostras (raspagens de amígdalas, fluidos orais, fluidos de processamento). Recolhemos sempre amostras nos mesmos locais do pavilhão, espalhados espacialmente por toda a instalação, para tentar obter a maior representação possível de todo o local. Recolhemos até 16 amostras por local, todos os meses, que foram analisadas por PCR quantitativo, tendo a ORF5 sido sequenciada nas amostras positivas.
O que é que descobrimos?
A nossa descoberta mais importante foi que, em condições de campo, fomos capazes de detetar até três linhagens diferentes de vPRRS durante um único evento de amostragem em explorações de criação e até duas em explorações de engorda (Figura 2).
Figura 2. Sublinhagens de vPRRS encontradas nas explorações de estudo (1-5) ao longo da duração dos nossos eventos de amostragem (em meses) para os diferentes tipos de amostras.
| Evento de amostragem | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Exploração | Tipo de amostra | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 |
| 1 | Fluido de processamento | L1H | L1H | ||||||||||
| Raspagem das amígdalas | |||||||||||||
| 2 | Fluido de processamento | L1H | L1H | L1A | L1H | L1H | |||||||
| L1H | |||||||||||||
| L8† | |||||||||||||
| Raspagem das amígdalas | L1H | ||||||||||||
| 3 | Fluido de processamento | L1H | L1H | L1H | L1H | ||||||||
| Raspagem das amígdalas | |||||||||||||
| 4 | Fluidos orais | L5† | L1A | ||||||||||
| L5† | |||||||||||||
| Raspagem das amígdalas | L1A | L5† | L5† | L1A | |||||||||
| L5† | |||||||||||||
| 5 | Fluidos orais | L1A | L1A | L5† | |||||||||
| Raspagem das amígdalas | L1A | L1A | |||||||||||
Também observámos que, no caso do nosso estudo, as amostras de fluido oral foram muito difíceis de sequenciar, enquanto os fluidos de processamento foram os mais fáceis. Considerando que ambas são amostras "compostas", este facto pode ser explicado pelos valores Ct mais elevados, que indicam uma menor quantidade de vírus nos fluidos orais em comparação com os fluidos de processamento. Tentámos realizar WGS, mas acabámos por não conseguir obter sequências completas do genoma de nenhuma das nossas amostras. Suspeitamos que tal possa dever-se aos tipos de amostras utilizadas, mas é necessária mais investigação sobre este tema.
Estes resultados demonstram a importância de considerar a sequenciação de várias amostras aquando da tomada de decisões importantes, embora compreendamos que tal implicará custos mais elevados.
Agradecimentos
O National Pork Board financiou este projecto. Gostaríamos de agradecer aos veterinários e produtores que foram fundamentais na recolha de amostras.


