

O síndrome reprodutivo e respiratório suíno (PRRS) continua a ser, de longe, a doença com maior impacto económico na indústria suína e o seu controlo está longe de ser satisfatório. Uma melhor e mais completa imagem da variação do vírus PRRS e a monitorização da circulação de novas estirpes dentro de uma determinada área/país/continente certamente ajudaria veterinários e produtores a implementar programas de controlo e, possivelmente, de erradicação. Por este motivo, a partir do final de 1990, a sequenciação do vírus PRRS começou a estar disponível em todo o mundo, principalmente na América do Norte, Europa e sudeste da Ásia.
O genoma do vírus PRRS (imagem 1) consiste numa molécula de ARN monocatenário que o torna propenso a "cometer erros" (mutações genéticas) durante a sua replicação no hóspede. Esta "tendência a cometer erros" resulta na presença, em campo, de diferentes estirpes de vírus PRRS, todas únicas na sua própria sequência genética. Entre os veterinários de campo e os investigadores continua a haver um debate sobre se estas diferenças nas sequências contribuem para "comportamentos diferentes" (sejam clínicos, patológicos ou imunológicos).

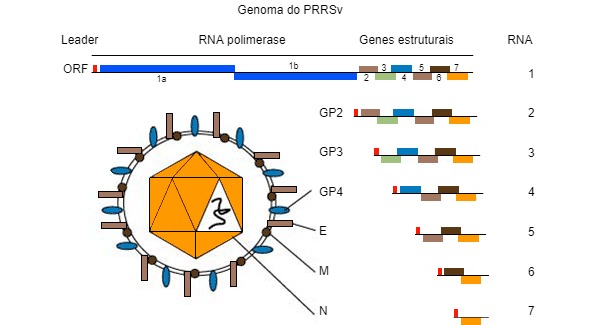
Figura 1. O genoma do vírus PRRS é uma molécula de ARN monocatenário.
Conceitos básicos da sequenciação do PRRS
A sequenciação do vírus realiza-se a partir de produtos de PCR procedentes de amostras de campo (soros, tecidos, fluídos orais) obtendo a leitura de nucleótidos geralmente de alguns fragmentos do genoma de ARN viral (ver figura 2) em determinadas regiões objectivo - ORF (Open Reading Frame) e depois são comparadas na percentagem de homologia, através da análise filogenética realizada recorrendo a softwares específicos. O resultado deste processo proporciona o grau de semelhança (homologia) entre as diferentes estirpes de vírus PRRS. Usando softwares de visualização gráfica, também pode ser obtido um dendograma (ou "árvore filogenética") que mostra o grau de relação com a sequência do vírus de referência (ver figura 3).

Figura 2. A sequenciação do vírus é realizada a partir de produtos de PCR obtendo a leitura de nucleótidos geralmente de alguns fragmentos do genoma do ARN viral em determinadas regiões objectivo- ORF.
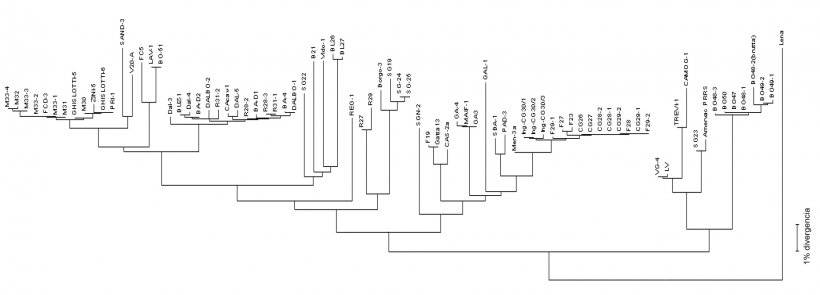
Figura 3. Os dendrogramas ou "árvore filogenética" são utilizados para representar gráficamente o grau de semelhança (homologia) entre diferentes virus PRRS com uma sequência de vírus de referência.
O genoma do PRRSV codifica, pelo menos, 10 ORF. Os mais frequentemente utilizados para a sequenciação - ainda que representem apenas 4% e 3% respectivamente do genoma completo, são ORF5 (que codifica a proteína E não glicosilada) e ORF7 (que codifica a proteína de nucleocápside (N)). A ORF5 representa uma região mais variável, enquanto que a ORF7 representa uma região mais conservada. Devido a isto, o mesmo grau de variação (por exemplo uma variação de 5%) que se encontre em ORF7 é mais "dramática" em termos de mudança genética, em comparação com a mesma variação em ORF5. A interpretação das semelhanças (ou seja, se os vírus estão relacionados ou não) requer muito mais informação adicional, já que a taxa de mudança genética pode ser muito variável.
É extremamente importante manter um arquivo de registo de todas as sequências identificadas univocamente, anotando cuidadosamente a data, o tipo de exploração (sítio 1-2-3), o fluxo suíno, a localização (latitude / longitude do GPS) e a origem da sequência (tipo de animal/tecido/amostra). Actualmente, a nossa base de dados de sequências de vírus PRRS abarca mais de 1300 sequências de ORF7 desde 2002. Para interpretar e dar sentido às diferenças, é ainda mais importante relacionar sequências individuais com eventos clínicos como o número de porcas que abortaram e a mortalidade pré.desmame nos Sítios 1 ou a taxa de mortalidade nos sítios 2 e 3.
Perguntas práticas
As perguntas frequentes de produtores e veterinários são:
- As diferenças genéticas observadas entre as sequências representam a variação normal de apenas uma estirpe de PRRS numa exploração/sistema, ou representan múltiplas estirpes diferentes presentes ao mesmo tempo, ou em curto espaço temporal, numa exploração/sistema?
- O que me está a acontecer agora é um "novo surto" causado por uma "nova estirpe" ou é uma recirculação?
Para responder a estas perguntas há que recordar o grau aceite de homologia de duas estirpes virais recolhidas dentro de um certo periodo de tempo (12-24 meses?). Por outras palavras, o ponto de corte de semelhança. Cerca de 97-98% de homologia na sequência (ou uma diferença de 2-3%) é um valor geralmente aceite. Segundo a minha experiência, é bastante difícil ver uma mudança superior a 2% numa "população fechada e clinicamente estável" (uma população convencional de porcas ou um fluxo de porcos) já que o observado é que obtemos a "mesma estirpe" durante um periodo de até 3 anos numa população única clinicamente estável. No polo oposto, cada vez que notamos uma actividade consistente de PRRS, recupera-se uma estirpe "nova" e filogeneticamente diversa (90% de homologia ou menos). Infelizmente, não se sabe com certeza se estas grandes diferenças, ás vezes observadas, são o resultado de uma mudança repentina no vírus/mutação (improvável, na minha opinião pessoal) ou a introdução de uma nova estirpe. O que está claro e bem aceite é que a semelhança/diversidade genética não é de nenhuma maneira indicativa quanto à semelhança imunológica (ou seja, indicativa de imunidade cruzada protectora) e não é de todo indicativa da patogenicidade intrínseca (não prevê se uma estirpe é particularmente "boa" ou "má").
As sequências completas do genoma que estão disponíveis na actualidade (infelizmente ainda mais para fins de investigação que para o uso diagnóstico diário) vão, sem dúvida, ajudar a responder esta pergunta.
É muito importante analisar as novas sequências de vírus PRRS contra um amplo conjunto de referência que represente a exploração, o sistema e a região, assim como as sequências das vacinas comerciais disponíveis (isto permitirá diferenciar entre estirpes de campo e de vacinas). Neste momento ainda está a ser usado um software de código aberto administrado pela Universidade de Pádua para construir as nossas árvores filogenéticas e manter a sua organização por fluxo de porcos dentro do total do nosso sistema de produção, num futuro próximo poderiam unir-se outros dois programas informáticos "ad hoc" (Bioportal da Universidade de Davis-California e CLASSIFARM-PATH de IZSLER (Bréscia, Itália) que terão um conjunto muito maior de sequências para comparar e permitir uma melhor compreensão do vírus PRRS circulante na Itália e possivelmente na UE.
Agradecimentos: Obrigado à Prof. Michele Drigo (UNI-PD) pela interessante discussão e a revisão deste documento.



")
