

Introdução
O genoma do virus do Sindroma Reprodutivo e Respiratório Suíno (PRRSV) é formado por uma cadeia simples de RNA, o que o torna muito propenso a mutações genéticas. Isto também faz com que cada estirpe de PRRSV seja única, pelo que a tipagem genética seja um método útil para o diagnóstico e para o controlo da doença. O diagnóstico por tipagem genética faz-se determinando a ordem dos nucleótidos dentro de uma cópia de DNA de um fragmento do genoma RNA – mediante sequenciação de DNA. Actualmente o fragmento mais utilizado é o ORF5, o gene que codifica para a glicoproteína mais importante da capsula, principalmente porque apresenta uma grande diversidade genética.

Diagnóstico por sequenciação de DNA
A discriminação entre o PRRSV tipo 1 (Europeu) e tipo 2 (Americano) faz-se facilmente com a maioria dos PCR diagnósticos, mas a discriminação entre estirpes individuais dentro de cada um dos dois genótipos requer sequenciação de DNA. Para isso, utilizam-se fluidos corporais ou tecidos, com uma carga entre moderada e severa de PRRSV, dos que se isola o RNA, que é copiado para DNA mediante transcrição inversa; então amplifica-se o gene ORF5 mediante PCR e envia-se para o laboratório de sequenciação de DNA. Este processo está muito automatizado e requer entre um e três dias. Os dados em bruto da sequenciação são enviados para o laboratório de diagnóstico para análise. O relatório costuma incluir a sequência de nucleótidos da estirpe, a sua similitude com estirpes vacinais standard e alguns laboratórios ainda fornecem uma comparação com um painel de referência standard de isolados selvagens de PRRSV em forma de dendrograma ou a comparação com a base de dados de PRRSV de um sistema de produção.
Analise de sequências de PRRSV
A semelhança, ou identidade, entre sequências determina-se alinhando duas ou mais sequências utilizando um programa informático. A figura 1 representa um exemplo de alinhamento de sequências de ORF5 de 5 explorações distintas. As comparações por pares da percentagem de identidade apresentam-se na tabela. As identidades oscilam entre 81,2 e 99,8 %. o dendrograma resultante da análise filogenética mostra um agrupamento das sequências similares (figura 2). Uma questão chave para os produtores e veterinários é se as diferenças genéticas observadas entre sequências representam uma variação normal de uma mesma estirpe de PRRSV da exploração ou representam várias estirpes distintas na mesma exploração.

Figura 1: Fragmento do alinhamento de sequências ORF5 de estirpes de PRRSV de 5 explorações distintas. Das explorações 1, 4 e 5 obtiveram-se múltiplas sequências. Os pontos representam posições idênticas à estirpe de referência: um PRRSV tipo 1, Lelystad.
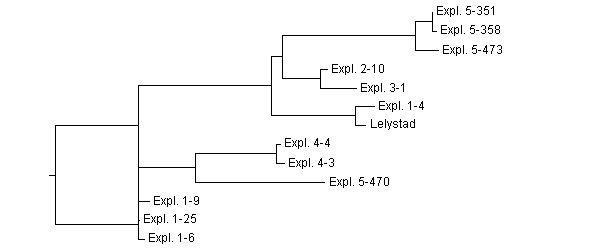
Figura 2: Dendrograma de sequências ORF5 obtidas de 5 explorações distintas. Exemplo de interpretação: na exploração 1 há duas estirpes não relacionadas. Três sequências são > 99 % idênticas entre si, enquanto que a quarta só está relacionada ~ 83 %. Pelo contrário, está relacionada com o vírus Lelystad. As estirpes da exploração 2 e 3 estão muito relacionadas (98,2 % idênticas). Duas estirpes da exploração 4 estão muito relacionadas (99,5 % idênticas). Na exploração 5 há duas estirpes distintas: três sequências são > 98 % idênticas entre si e 81 % com a quarta.
Tabela: Percentagem de identidade por pares entre todas as sequências alinhadas de ORF5 de uma série de amostras de PRRSV tipo 1.
| 1-4 | 1-6 | 1-9 | 1-25 | 2-10 | 3-1 | 4-4 | 4-3 | 5-351 | 5-358 | 5-470 | 5-473 | Lelystad | |
| *** | 83,5 | 83,3 | 83,7 | 93,2 | 92,6 | 86,1 | 86,0 | 88,1 | 88,0 | 83,2 | 88,3 | 98,7 | 1-4 |
| *** | 99,2 | 99,7 | 84,2 | 83,0 | 86,0 | 86,0 | 81,4 | 81,2 | 86,3 | 81,4 | 82,1 | 1-6 | |
| *** | 99,5 | 84,5 | 83,3 | 85,6 | 85,6 | 81,4 | 81,2 | 86,5 | 81,4 | 81,9 | 1-9 | ||
| *** | 84,5 | 83,3 | 86,0 | 86,0 | 81,7 | 81,5 | 86,8 | 81,7 | 82,2 | 1-25 | |||
| *** | 98,2 | 86,6 | 86,5 | 91,1 | 90,9 | 84,2 | 90,4 | 93,3 | 2-10 | ||||
| *** | 84,4 | 84,2 | 90,2 | 90,0 | 83,3 | 89,7 | 92,9 | 3-1 | |||||
| *** | 99,5 | 82,5 | 82,7 | 90,6 | 82,5 | 84,8 | 4-4 | ||||||
| *** | 82,3 | 82,5 | 90,9 | 82,2 | 84,6 | 4-3 | |||||||
| *** | 99,8 | 81,0 | 98,3 | 88,4 | 5-351 | ||||||||
| *** | 81,2 | 98,2 | 88,2 | 5-358 | |||||||||
| *** | 80,9 | 83,2 | 5-470 | ||||||||||
| *** | 88,8 | 5-473 | |||||||||||
| *** | Lelystad |
Interpretação das sequências de PRRSV
A sequência de ORF5 tem uns 600 nucleótidos. Diversas estimativas indicam que a taxa global de mutação deste gene é, aproximadamente, de 0,5 - 1 % por ano. As variações na taxa de alteração genética são determinadas por distintos factores não víricos. O nível de imunidade específica contra o PRRSV dos porcos tem um grande impacto na replicação e na transmissão viral, exercendo uma forte pressão inibidora que diminui o número de cópias. Uma carga viral reduzida comporta uma menor taxa de transmissão, limitando todavia mais a replicação viral e diminuindo a taxa de alteração. O mesmo vírus pode apresentar distintas taxas de alteração genética sob distintas condições pelo que, nalguns casos, podem observar-se taxas de alteração maiores ou menores que o intervalo sugerido de 0,5 - 1% por ano.
A questão principal na análise genética é saber se duas sequências estão muito relacionadas (pertencem a duas variantes da mesma estirpe) ou são independentes (pertencem a duas estirpes não relacionadas). Comummente aceita-se que dois isolados de PRRSV estão relacionados ou não se a sua similitude está acima ou abaixo de 97 - 98 %. È evidente que basear-se unicamente numa diferença genética de 2 ou 3 % entre dois isolados, sem mais informação adicional, pode levar a conclusões erradas. As diferenças entre variantes de uma mesma estirpe circulante numa população durante vários anos podem exceder esta cifra. A interpretação da relação será melhor se se tem acesso a informação adicional, incluindo as datas e lugares dos isolamentos. É muito importante comparar as novas sequências de PRRSV com um amplo grupo de referência que seja representativo da exploração, do sistema e da regíão e que também represente a diversidade genética global.
A sequenciação de DNA de cadeias de PRRSV pode indicar uma relação de proximidade, ou de independência, das cadeias (figura 2) mas não pode prever ou explicar a protecção imunológica ou os focos de PRRS em explorações imunes. Tampouco permite a previsão da evolução clínica da infecção de uma estirpe concreta, já que os marcadores genéticos de virulência ainda não se identificaram.
Actualmente há muitos projectos regionais em marcha de controlo ou eliminação do PRRS, especialmente nos EUA, mas também na Europa. Ter uma visão completa da variabilidade viral no início do projecto de controlo e eliminação do PRRS é essencial para um acompanhamento eficaz do progresso e da eficácia dos processos implementados e para identificar os novos vírus introduzidos nas explorações da região.




")